![]()
![]()
![]()
von Marc Niggemann
Dies ist eine Facharbeit, die ich im Rammen eines zweiwöchigen Praktikums am Wilhelm-Jost-Institut (Hamm) erstellt habe. Die Facharbeit wurde in der Oberstufe meiner Schule als Klausurersatz gewertet.
Inhaltsverzeichnis
Seite
|
1. Einleitung 2. Stoffuntersuchung von Methanol mit dem Spektrometer 2.1 Versuchsvorbereitung und Aufbau für die Untersuchung von Methanol 2.2 Hauptuntersuchung mit dem Spektometer 2.2.1 Auswertung der Kurve von Methanol 2.3 Vergleich mit einer optimalen Normkurve 2.4 Fehlerdiskussion in Bezug zur Versuchsmessung von Methanol 3. Diskussion über Fehler die bei der einfachen Stoffuntersuchung auftreten können, ihre Ausmaße sowie ihre Vermeidung 3.1 Unreine Küvetten 3.2 Zu dicke Proben und zu hohe Sensibilität 3.3 Der Einfall von Streulicht 3.4 Warmlaufen des Spektrometers 3.5 Gase oder Partikel in der Umgebungsluft 3.6 Flüchtige Stoffe 4. Analyse von Stoffgemischen 5. Quantitative Untersuchung 6. Literaturverzeichnis 7. Bildverzeichnis 8. Anhang 8.1 Technisch-pysikalische Grundlagen für das Infrarot- Spektrometer 8.2 Grundprobleme bei der IR-Spektroskopie 8.3 IR-Spektren 8.4 Stray light and I0 line 8.5 Model x98 Block Diagram 8.6 Typical Spectral Slit Width Curve 8.7 Tabelle A.135 Charakteristische Gruppen- und Gerüstfrequenzen im IR-Gebiet
|
3
4 5 6 6 7
8 9 9 10 10 11 11 12 15 16 17 19 21 39 40 41 42 |
Einleitung
Diese Facharbeit behandelt das Thema der Ungenauigkeit von Spektrometern, insbesondere des x98 Perkin-Elmer Spektrometers. Im ersten Teil dieser Arbeit wird die Vorgehensweise an einer beispielhaften Untersuchung beschrieben, um eine allgemeine Vorgehensweise als Grundlage zu haben. Im darauf aufbauenden zweiten Teil werden die einzelnen Fehlerquellen erläutert, die zu dem ungenauen Ergebnis beitragen. Das Schwerpunktthema ist die Vermeidung und Minimierung der festgestellten Fehlerquellen. Zudem werden Auswirkungen von bestimmten Fehlern, die zu einer extremen Fehlmessung führen, an anderen Meßbeispielen als gesondertes Thema behandelt. Der dritte Teil behandelt die Problematik bei der Analyse von Stoffgemischen und der quantitativen Analyse. Im Anhang werden die technischen und physikalischen Grundlagen für das technische Prinzip des allgemeinen Spektrometers kurz erläutert. Im Hauptteil ist bewußt auf die Erklärung von Grundlagen verzichtet worden, um das Thema auf das Wesentliche beschränken zu können. Im Anhang finden sich Messungen von 4 Stoffen und die dazugehörigen Normkurven. An ihnen kann die Genauigkeit des Gerätes selbst beurteilt werden.
2. Stoffuntersuchung von Methanol mit dem Spektrometer
2.1 Versuchsvorbereitung und Aufbau für die Untersuchung von Methanol
Methanol hat aufgrund seiner molekularen Struktur sehr charakteristische Peaks zwischen 1600 bis 800 cm-1 (Wellenzahl). Um diese messen zu können, müssen KBr-Platten verwendet werden. Sie absorbieren erst ab einer Wellenzahl von kleiner als 400 cm-1. Die folgende Messung soll als reine Identifikationskurve für den Stoff gelten, deshalb wird eine Küvette verwendet (Schema 1, Foto 1), die einen dünnen
Kapillarfilm der Flüssigkeit zwischen zwei KBr Platten bilden kann. Der Abstandhalter von 0,025 mm läßt eine Messung zu, die die Peaks nicht bis ins Unendliche ausschlagen lassen, und so die schwächeren und auch die stärkeren Peaks anzeigen. Eine dickere Schicht als 0,1 mm absorbiert zu stark und die Peakspitzen sind in dieser Messung, außerhalb des Meßbereiches. Die zusammengebaute Küvette wird darauf in das Spektroskop eingehangen, um die Basislinie aufzuzeichnen. Falls die Küvette zu lange der Luft ausgesetzt wird, ist die obere Schicht der KBr Platten angelöst (Salzlösung) und die Basislinie nach unten verschoben, weil die nötige Durchlässigkeit nicht mehr gegeben ist. Zudem erkennt

man vorzeitig Verunreinigungen, die bei der Analyse nur mit Mühe festzustellen sind und die Analyse unbrauchbar machen (darauf wird in den nachfolgenden Kapiteln näher eingegangen).
Als Letztes muß die Sensibilität des Gerätes dem Stoff angepaßt werden. Hierzu prüft man durch die Unterbrechung der Lichtquelle, vor der Probe, die 100%-ige Absorption des
Lichts. Die Plotternadel schlägt darauf hin aus. Die Geschwindigkeit des Anzeigerausschlages sollte bei dieser Analyse im mittleren Bereich sein. Dadurch werden dann kleinere Schwingungen nicht unterdrückt und das "Hintergrundrauschen" wird eingeschränkt.
2.2 Hauptuntersuchung mit dem Spektrometer
Nachdem die Voreinstellungen des Spektrometers abgeschlossen sind, wird die Probe in den Meßschacht eingehängt. Sollten die ersten Peaks schon an der Unendlichlinie anschlagen, ist es möglich, während der Messung die Basislinie mehr nach oben zu verstellen und die Sensibilität zu verringern. Der Sprung in der Messkurve sollte schon während der Messung markiert werden, um den späteren Meßfehler eindeutig lokalisieren zu können.
In dieser Messung von Methanol ist die Voreinstellung ausreichend gewesen. Die Nachjustierung hätte die Kurve zu sehr verändert um sie als Eichkurve benutzen zu können. Der Scan hätte in diesem Fall abgebrochen werden müssen um mit einer neuen Voreinstellung erneut zu beginnen.
2.2.1 Auswertung der Kurve von Methanol
Um die Peaks zuordnen zu können, wird zunächst den Peaks die Wellenlänge zugeordnet. Hierzu muß ein "Maßband" angelegt werden, daß die logarithmische Aufteilung wiedergibt. Es ist auch möglich, daß während der Messung die Peaks anhand der Anzeige des Spektrometers mit einem Stift markiert werden. Der Stoff in diesem Versuch ist bekannt (Schema 2), und so können die Peaks mit Hilfe der Tabelle "charakteristische Gruppen- und Gerüstfreqenzen im IR
- Gebiet" (siehe Anhang) identifiziert werden.
Der Peak 1 (Abb. 20, Methanol) ist als O-H Valenz zu erkennen, da er zwischen 3600-3200 cm-1 liegt. Eine N-H Valenz ( 3550- 3350 cm-1 ) kann es nicht sein, weil kein N in der Summenformel (Schema. 2) vorhanden ist.
Peak 2: C-H-Valenz (3000-2800 cm-1)
Peak 3: CH2 -Valenz (2925-2850 cm-1 )
Peak 4: CH3-Deform (1390-1370 cm-1)
Nun läßt sich schon feststellen, daß kein Fremdstoff in der Probe des Methanol gewesen ist, weil keine Peaks vorhanden sind, die nicht zu zuordnen sind.
2.3 Vergleich mit einer optimalen Normkurve
Bei dem Vergleich der Kurve aus dem Versuch 1 und der Normkurve (Abb. 13) von Methanol (Abb. 20) ist der Kurvenverlauf identisch. Es läßt nun mit Sicherheit sagen, daß kein Fremdstoff in der Probe gewesen ist.
Die markanten Stellen sind nachträglich auf der Normkurve markiert (1-9) und lassen sich auch alle auf der Versuchskurve wiederfinden. Der Ausschlag der Peaks kann auf den beiden Diagrammen unterschiedlich groß sein, da dies an dem Verstärkungsfaktor des Spektrometers, sowie an der eingestellten Sensibilität oder an der Schichtdicke der Probe liegt. Im vorliegenden Fall sind die Peaks der Versuchskurve eindeutig stärker, aber dennoch lassen sich kleine Merkmale erkennen. Die sehr schwachen Peaks 1 und 6 (siehe Diagram 20, Methanol), sind trotz der relativ hohen eingestellten Sensibilitat und dem dadurch begünstigten "Hintergrundrauschen", gut zu erkennen. Bei dem Peak A ist ein leichte Abflachung zu erkennen die nicht in der Normkurve dargestellt ist.
Im Gegensatz zur Stärke der Peaks, muß die Lage der Peaks genau sein, da man sonst die Zuordnung nicht genau durchführen kann. Dazu werden markante Punkte wie die Spitze der Peaks mit der Wellenzahl gekennzeichnet (die meisten Punkte können schon von der Bestimmung verwendet werden). Bei der Versuchskurve (Abb. 20) ist bei 3000 cm-1 ein Wellenberg, aber bei der Normkurve ist diese Stelle bei ca. 3050 cm-1. Der Punkt 4 endet bei 2500 cm-1 und stimmt so mit der Versuchskurve überein. Auch der Peak bei 1025 cm-1 ist bei beiden Kurven identisch.
Im Abschnitt von 700 - 400 cm-1 allerdings ist das Ergebnis verfälscht und nicht annähernd so wie es zu erwarten wäre (dies ist auch bei weiteren Messungen von andern Stoffen aufgetreten).
Abschließend läßt sich sagen, daß in diesem Versuch alle markanten Punkte vorhanden sind, und daß die Wellenzahl im Teil von 2500-1000 cm-1 genau ist. Der Wellenzahlbereich davor hat allerdings eine Ungenauigkeit von -50 cm-1. Der Bereich von 700-400 cm-1 ist so ungenau, daß er nicht zur Bewertung des Stoffes herbeigezogen werden kann.
2.4 Fehlerdiskussion in Bezug zur Versuchsmessung von Methanol
Wie schon in dem vorherigen Abschnitt erwähnt, gibt es keine Fehlpeaks, die nicht dazugehören. Dennoch ist an dem Peak A eine Abflachung zu erkennen (Abb. 20), die in der Normkurve als Rundung zu sehen ist. Dieser Fehler kann damit erklärt werden, daß der Bereich in der Wellenzahl liegt, wo das Spektrometer sich neu ausrichtet. Dabei ändert es die Geschwindigkeit des Scanvorganges (zum Ende wird sie verlängert) und gibt so genauere Daten (höhere Auflösung) für z.B. Aromate, die im Endbereich markante Peaks haben. Es kann geschehen, daß in dieser Phase Meßfehler auftreten, und so kann diese Abweichung von der Normkurve erklärt werden.Der Bereich zwischen 700-400 cm-1 ist, wie schon genannt, unbrauchbar, und ist auf innere Fehler des Spektrometers zurückzuführen. Vermutlich ist das Prisma, daß für den Bereich zuständig ist oder der Detektor, der die Strahlung in Impuls umwandelt, beschädigt oder verunreinigt.
3 Fehlerquellen, die bei der einfachen Stoffuntersuchung auftreten können, ihre Ausmaße sowie ihre Vermeidung
3.1 Unreine Küvette
Der erste Fehler kann schon mit einer unreinen Küvette (Foto 2) anfangen. Dieses äußert sich darin, daß Fremdsubstanzen mitgemessen werden können (Abb. 1) oder, daß die Basislinie nach unten verschoben ist, da zuviel Strahlung von den Fenstern selbst absorbiert wird.
Bei KBr Fenstern, die Verunreinigungen oder Riefen sowie Ausspülungen aufweisen, kann die Oberfläche mit feinem Schleifpapier (800 Körnung) abgeschliffen werden und anschließend mit Poliermittel poliert werden. In der Regel werden die Fenster danach wieder sehr klar. Die KBr-Fenster sind sehr empfindlich. Wenn sie zu fest in die Halterung montiert werden, können sie brechen. Kleinere Risse oder Sprünge beeinträchtigen die Messungen nicht. Es ist aber möglich, daß durch solche Risse die leicht flüchtigen Stoffe schneller entweichen.
3.2 Zu starke Proben und zu hohe Sensibilität
Ein weiterer Fehler, der mit der Probenvorbereitung zusammenhängt, ist die Verwendung von zu dicken Abstandhaltern (Zeichnung. 1) in der Küvette. Das hat zur Folge, daß Peaks sehr stark ausschlagen und an die ¥ -Grenze anschlagen (Abb. 28). Dieses kann auch daran liegen, daß die Sensibilität zu hoch eingestellt ist.
3.3 Der Einfall von Streulicht
Die meisten Spektrometer haben eine Klappe (Foto. 3) , die den Meßschacht gegen Streulicht aus dem Labor abschottet. Zudem arbeiten sie mit einer Lochblende. Sie sorgt dafür, daß nur Licht verstärkt zu einen Signal umgewandelt wird das von der eigenen Lichtquelle stammt. Zwei Messungen mit dem gleichen Stoff und den gleichen Einstellungen ergab, daß kein wesentlicher Unterschied zu erkennen ist.
Lichtquellen die IR-Licht abgebe werden normaler weise nicht zur Beleuchtung benutzt. Licht welches durch die Fenster einfällt, kann keine infrarote Licht mehr enthalten, weil es wie die Glas Küvetten, infrarote Strahlung absorbirt. Die Beleuchtung mit Neonröhren strahlt grundsätzlich wenig infrarotes Licht ab. Glübirnen geben sehr viel dieser Strahlung ab. Sie aber haben einen Glaskolben der einen großen Teil ihrer Strahlung absorbiert. Deswegen ist der Einfall von Streulicht auszuschließen oder im ungünstigsten Fall vernachlässigbar gering.
3.4 Warmlaufen des Spektrometers
In der Gebrauchsanleitung zu dem Spektrometer wird geraten das Gerät ca. 20 min. warmlaufen zu lassen, bevor man mißt. Die Lampe erreicht erst dann ihre richtige Temperatur und bekommt so ihre ausreichende Helligkeit. Zu frühe Messungen führen zu einer flacheren Kurve.
Die Messung von Methanol ohne "warmlaufen" und ohne langes Einstellen, ergab eine normale Kurve (Abb. 5). Eine zweite Messung nach den angegebenen 20 min. Wartezeit ergab fast dieselbe Kurve mit nur wenigen Unregelmäßigkeit in den beiden Kurven. Diese Unterschiede sind zu vernachlässigen (siehe Vergleichspunkt in Kurve 5 und 6)
3.5 Gase oder Partikel in der Umgebungsluft
Das Spektrometer verfügt über einen parallelen Meßschacht, mit dem Lösungsmittel aus einer Messung herausgefiltert werden können, wenn eine Reinprobe des Lösungsmittels in den zweiten Schacht eingehängt ist. Das Spektrometer registriert dann nur Unterschiede zwischen den beiden Schächten.
Dieser Mechanismus ist ständig in Betrieb und sorgt dafür, wenn kein Lösungsmittel gebraucht wurde, daß die Luft vor dem Detektor und der Lampe nicht mitgemessen wird. Wenn die Verteilung von Gasen oder Partikeln unterschiedlich sein sollte, ist in beiden Schächten nicht mehr dasselbe Gemisch und die Differenz wird angezeigt, welche die Messergebnisse verfälschen würde.
Eine Möglichkeit, um die Ausmaße einer solchen Ungenauigkeit festzustellen, ist die Verdrängung der Luft mit einem reinem Gas. Dieses würde sich im Schacht ausbreiten und ihn schließlich füllen. Ein Problem ist allerdings, daß der Schacht (Foto. 12) nicht dicht ist, weil dort Öffnungen zu der Lampe sowie zu dem Detektor im Spektrometer eingelassen sind.
CO2 ist für den Zweck ein sehr gut geeignetes Gas, weil es schwerer als Luft, nicht giftig und auch nicht brennbar ist. Bei der Einleitung mit einem Schlauch in den Schacht wird die Luft verdrängt, und das CO2 flutet langsam durch das ganze Gerät. Wegen der Verflüchtigung durch Lüftungsschlitze und Verwirbelung an der Grenzschicht zu Umgebungsluft muß ein leichter Zustrom von CO2 ständig während der Messung fließen.
Die Messung von Methanol (Abb. 20-A) ergab kein überragendes Ergebnis, da das Hintergrundrauschen nicht beeinflußt worden ist. Es ist davon ausgegangen worden das mindestens ein Teil des Hintergrundrauschen von den Verunreinigungen in der Luft abhänig ist Beim Vergleich mit einer Kurve, die ohne CO2 Flutung gemacht worden ist sind kleine Unebenheiten verschwunden (siehe Vergleichspunkte Abb.20).
Der technische Aufwand und die Kosten sind verhältnismäßig hoch und nur für sehr genaue Messungen zu empfehlen.
3.6 Flüchtige Stoffe
Das Problem bei sehr flüchtigen Stoffen ist, daß sie nicht lange genug in der Küvette bleiben bis die Messung zu Ende ist. Das liegt hauptsächlich daran, daß sich die Flüssigkeit durch die Absorption der Strahlung erwärmt und so die Flüssigkeit verdampft. Durch Stopfen, die die Einfüllstutzen verschließen, läßt sich die Verdampfung hinauszögern und es reicht aus, wenn man die Scan-Zeit auf 3 min. stellt. Falls es nicht reicht, wird nur der erste Peak normal aufgezeichnet und danach folgt eine gerade Linie (Abb. 27). Bei der Verwendung eines sehr flüchtigen Stoffes als Lösungsmittel, kann dieser Stoff aus der Lösug entweichen. Die Verflüchtigun wird während der Untersuchung, durch die Erwährmung von der Strahlung gefördert und verfälscht die Analyse.
4. Analyse von Stoffgemischen
Bei der Analyse von Stoffgemischen ergibt sich eine Überlagerung der einzelnen Kurven. Bei Peaks, die von der Wellenzahl übereinanderliegen ergibt sich ein gemeinsamer Peak.
Falls in dem Diagramm eines Stoffes 1 zwischen 2500-2000 cm-1 ein Peak vorhanden ist, und nun ein weiterer Stoff 2 zugegeben wird, der nur bei 800 cm-1 einen Peak aufweist, ergibt sich ein neues Diagramm mit zwei Peaks bei 2500-2000 cm-1 und 800 cm-1. Um zu sehen, in welcher Konzentration ein Stoff noch Einfluß hat, ist eine Versuchsreihe zu messen, in der die Konzentration von einem Stoff sinkt.
Die Stoffe müssen sich zudem vermischen und keine Grenzschichten bilden. Dafür ist Methanol und Essigsäure geeignet .
Die erste Messung mit einem Mischungsverhältnis von Essigsäure zu Methanol von 1:1 (Abb. 21) ergab eine zu erwartende Mischkurve (vergleiche Reinsubstanzen Abb. 20, 22).
Die nächsten Mischungen von 1:2 und 1:4 (Abb.15 und 16) ergaben keine rapide Abnahme. Erst bei einer Konzentration von 1:100 läßt sich eine eindeutige Abnahme erkennen (vergleiche Abb. 23-27). Die folgenden Konzentrationen von 1:200 und 1:300 zeigen die Abnahme der Essigsäure-Peaks gegen Null.
Bei einer Mischung Essigsäure zu Methanol von 1:400 lassen sich noch Spuren erkennen ,obwohl die Peaks von Methanol eindeutig dominieren.
Dies zeigt, daß selbst kleine Verunreinigungen noch Folgen haben.
5. Quantitative Untersuchungen.
Das Spektrometer mißt die Stärke der Absorption von einer bestimmten Wellenlänge eines Stoffes. So ist es theoretisch möglich, auch quantitativ zu messen. Das wird häufig gebraucht, wenn z.B. Reaktionsprodukte in einer Lösung bestimmt werden müssen. Falls die Produkte dann in einer sehr geringen Menge vorhanden sind und deswegen eine sehr starke Schicht des Stoffes zu untersuchen ist, entstehen daraus Probleme.
Meistens stehen nur offene Küvetten (Foto. 2) aus Glas zur Verfügung, die nicht bis in den langen Wellenbereich die Messung zulassen (Abb. 28). Der erste Bereich reicht eventuell für eine Identifikation aus. Um nun das reine Reaktionsprodukt zu erhalten, muß das Ausgangsprodukt, welches auch in der Lösung ist, durch den zweiten Strahlengang (Foto. 4) herausgefiltert werden.
Dies ist in der dünnsten Küvette unmöglich, denn das IR-Gerät schlägt immer bis zur Unendlichlinie aus. Die Reinprobe im zweiten Strahlengang hat kein Einfluß auf die starke Empfindlichkeit des Spektrometers. Mit anderen Arten des Spektrometers ist dieses jedoch möglich.
6 Literaturnachweis
Czieslik Wolfgang, Moderne Analysenmethoden Teil 2 Spektroskopische Methoden, Aulis Verlag Deubner und Co KG, Köln,1883
Götz Dieter, Moderne Analysenmethoden Teil I Elektroanalytische Methoden, Aulis Verlag Deubner und Co KG Köln,1883
Hannah R.W. / Swinehart J.S., Experimente zur Infrarotspektroskopie, Perkin-Elmer Corporation Infrarot-Anwendungslabor, Badenseewerk Perkin-Elmer und Co GmbH, Überlingen, o. J.
Kober Demuth, Grundlagen der Spektroskopie, Verlag Sauerländer AG, Salle, 1977
Microsoft, Encarta 97 Enzyklopädie, o. O, 1997
Morrison Robert T. / Boyd Robert T., Lehrbuch der Organischen Chemie, VCH Verlagsgesellschaft mbH, D-6940 Weinheim, 19863
Perkin- Elmer Ltd., Model x98 Infrared Spectrophotometer Operator`s Manual, Beaconsfield, Buckinghamshire, 1979
Schulze Gerhart / Simon Jürgen, Maßanalyse, Walter de Gruyter, Berlin und New York 198614
Schwedt Georg, Analytische Chemie, Georg Thieme Verlag Stuttgart / New York, o. O., 1995
7 Bildverzeichnis
Abbildungen: Seite
| 1 | Eigenanfertigung, Analyse von einer unreinen KBr | 22 |
| 2 | Eigenanfertigung, Analyse ohne einer KBr | 22 |
| 3 | Eigenanfertigung, Analyse von einer sauberen KBr | 23 |
| 4 | Eigenanfertigung, Analyse mit sehr hoher Sensibilität durch verlangerung der Scan Zeit auf 60 min. bez. 240 min. | 23 |
| 5 | Eigenanfertigung, Analyse von Methanol mit warmlaufen lassen des Spektrometers | 24 |
| 6 | Eigenanfertigung, Analyse von Methanol ohne warmlaufen lassen des Spektrometers | 24 |
| 7 | Eigenanfertigung, Heptan mit einer Scan zeit von 12 min. | 25 |
| 8 | Eigenanfertigung, Heptan mit einer Scan zeit von 6 min. | 25 |
| 9 | Eigenanfertigung, Heptan mit einer Scan zeit von 3 min. | 26 |
| 10 | Sadtler, Normkurve von 1-Hepten, Research Laboratories, Inc., Phiadelphia, 1975, | 27 |
| 11 | Sadtler Ebda, Normkurve von Hexan | 28 |
| 12 | Sadtler Ebda, Normkurve von Cyclohexan | 28 |
| 13 | Sadtler Ebda, Normkurve von Methanol | 29 |
| 14 | Sadtler Ebda, Normkurve von N-Heptan | 30 |
| 15 | Sadtler Ebda, Normkurve von Cyclopentan | 30 |
| 16 | Eigenanfertigung, Vergleichsmessung von Cyclhexan | 31 |
| 17 | Eigenanfertigung, Vergleichsmessung von N-Heptan | 31 |
| 18 | Eigenanfertigung, Vergleichsmessung von 1-Hepten | 32 |
| 19 | Eigenanfertigung, Vergleichsmessung von Cyclopentan | 32 |
| 20 | Eigenanfertigung, Vergleichsmessung von Metanol unter CO2 flutung | 33 |
| 21 | Eigenanfertigung, Mischung von Essigsäure zu Methanol im verhältnis 1:1 | 34 |
| 22 | Eigenanfertigung, Vergleichsmessung von Essigsäure | 34 |
| 23-A | Eigenanfertigung, Mischung von Essigsäure zu Methanol im verhältnis 1:4 | 36 |
| 23 | Eigenanfertigung, Mischung von Essigsäure zu Methanol im verhältnis 1:12 | 36 |
| 25 | Eigenanfertigung, Mischung von Essigsäure zu Methanol im verhältnis 1:100 | 37 |
| 26 | Eigenanfertigung, Mischung von Essigsäure zu Methanol im verhältnis 1:200 | 37 |
| 27 | Eigenanfertigung, Mischung von Essigsäure zu Methanol im verhältnis 1:400 | 38 |
| 28 | Eigenanfertigung, reines Methanol | 38 |
| 27-A | Eigenanfertigung, Flüchtiges Hexan | 39 |
| 28-A | Eigenanfertigung, Analyse mit zu starken Abstanzhaltern | 39 |
| 29 | Eigenanfertigung, Quantitaive Analyse | 40 |
|
Perkin- Elmer Ltd., Model x98 Infrared Spectrophotometer Operator`s Manual, Beaconsfield, Buckinghamshire, 1979 Figure 2.8 |
41 | |
| Perkin- Elmer Ltd Ebda., Figure 3.1 | 42 | |
| Perkin- Elmer Ltd Ebda., Figure 2.5 | 43 | |
| Unbekannt, Charakteristische Gruppen- Gerüstfrequenzen im IR-Gebiet | 44 | |
| Hannah R.W. / Swinehart J.S., Experimente zur Infrarotspektroskopie, Perkin-Elmer Corporation Infrarot-Anwendungslabor, Badenseewerk Perkin-Elmer und Co GmbH, Überlingen, o. J., Schema 1 | 45 |
8 Anhang
8.1 Technisch-pysikalische Grundlagen für das Infrarotspektrometer
Die infrarote Strahlung liegt im elektromagnetischen Spektrum zwischen dem sichtbaren Licht und der Mikowellenstrahlung (Abb 29). Das Spektrum selbst ist nach der Frequenz oder der Wellenlängeneinheiten der Strahlung geordnet.
Der Bereich der Infrarot-Strahlung wird aber üblicherweise in Wellenzüge pro cm (Wellenzahlen cm-1) angegeben. "Die Frequenz f und die Wellenlänge l sind durch die Gleichung [f * l = c] verbunden, wobei die Frequenz f die Schwingungen pro Sekunde und c die Lichtgeschwindigkeit (3 * 1010 cm/sec) bedeuten.
Eine Wellenzahleinheit n ist definiert als der reziproke Wert der Wellenlänge (n = 104 /l )." Der Grundschwingungsbereich, mit dem das IR arbeitet, liegt zwischen 4000 und 400 cm-1.
Es bestehen sämtliche Moleküle aus Atomen, die durch die chemischen Bindungen zusammengehalten werden (Schema 2. ). In der Bindung können die Atome schwingen und haben immer eine gleiche Frequenz. Wenn die IR-Strahlung nun die gleiche Frequenz wie z.B. die schwingenden Atomenden von einem Hexan Molekül besitzt, wird die Strahlung absorbiert. Die Energie des Moleküls wird erhöht, indem die Elekronen auf eine höhere Bahn gehoben werden (es wird erwärmt). Bei der IR- Analyse wird der genannte Effekt ausgenutzt, indem man die Probe mit infrarotem Licht durchleuchtet und die Wellenlänge von 4000 cm-1 bis auf 400 cm-1 hinunterregelt. Absorbiert z.B. ein Stoff die Strahlung etwa bei 1360 bis 1030 cm-1, kann man mit Hilfe von Vergleichskurven, die von bekannten Stoffen aufgenommen worden sind, eine C-N-Valenz Bindung charakterisieren.
Die IR- Analyse beschränkt sich auf einen kleinen Teil des Gesamtspektrum ( Abb. 29), aber dennoch reicht dieser Teil aus, um einen bekannten Stoff eindeutig zu identifizieren.
Bei dem Spektrum des Sonnenlichtes z.B. läßt sich so anhand der fehlenden Freqenzen (schwarze Striche) (Abb. 30), Materie in der Sonne oder um die Sonne herum, nachweisen.
Bei unbekannten Stoffen werden die Peaks mit Bekannten verglichen, dabei ist die IR-Analyse nur als Teil einer Gesamtanalyse zu sehen.
Abbildung 29 Übersicht über das elektromagnetische Spektrum
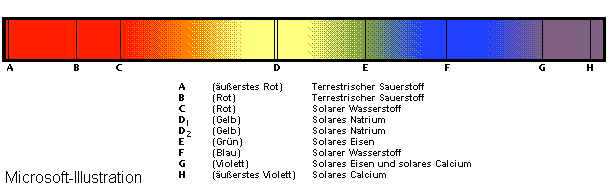
Abbildung 30: Spektrum des Sonnenlichts ( nur das sichtbare Licht )
8.2 Grundprobleme bei der IR-Spektroskopie
Die Fehler, die zu einem ungenauen Ergebnis führen, können schon entstehen wenn ein nicht reiner Stoff ausgewählt wird. Beispielsweise bei der Aufnahme einer Eichkurve, für die Bestimmung von Mengen in Lösungen. Bei einer qualitativen Analyse ist es wichtig, daß der Stoff möglichst frei von Fremdsubstanzen ist. Es sollte also die reinste zur Verfügung stehende Form der Stoffe verwant werden, die keine unbekannten Lösungsmittel oder Fremdstoffe enthalten, die man nicht durch den parallelen Meßschacht (Referenzprobe) herausfiltern kann.
Weiter sollte man darauf achten, daß sich die Küvetten durch die zu untersuchenden Stoffe wie Wasser, nicht auflösen, wenn man KBr -Platten (Foto 2) verwendet. Teilweise ist es ausreichend, die normalen Quarzküvetten zu verwenden, wenn die markanten Peaks in einem Bereich liegen in denenie Strahlung nicht absorbiert.
Für Testkurven, um die Tauglichkeit der Geräte zu prüfen, ist es nicht zu empfehlen, leicht flüchtige Substanzen wie Hepten zu verwenden. Diese Versuche führen oft zu Schwierigkeiten wegen der starken Verdunstungsrate. Die Proben in der Küvette werden von der Lichtquelle so stark aufgeheitzt, daß sie als Gas entweichen. Das hat zur Folge, daß nur ein kleiner Teil, ohne weitere technische Maßnahmen, analysiert werden kann. Daran läßt sich erkennen, daß es schon bei der Voruntersuchung der Stoffe liegt, ob das Ergebnis seinen optimalen Wert an Genauigkeit erreicht hat.
Ein Wort des Dankes...
...an alle Mitarbeieter des Wilhelm-Jost Institut, die mich betreut und unterstützt haben.